The Ab Initio Nanoreactor
Main content start
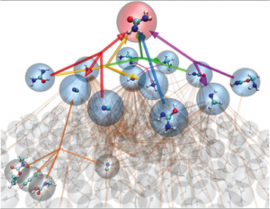
Chemical understanding is driven by the experimental discovery of new compounds and reactivity, and is supported by theory and computation that provide detailed physical insight. The ab initio nanoreactor is a highly accelerated first-principles molecular dynamics simulation of chemical reactions that discovers new molecules and mechanisms without preordained reaction coordinates or elementary steps. Using the nanoreactor, we can find new pathways which highlight the emergence of theoretical and computational chemistry as a tool for discovery.
Watch nanoreactor simulation videos on youtube here and here.
Related Publications
- Lee-Ping Wang, Alexey Titov, Robert McGibbon, Fang Liu, Vijay S. Pande, and Todd J. Martínez, Discovering chemistry with an ab initio nanoreactor, Nature Chemistry, 6, 1044-1048, 2014. [link]
- Lee-Ping Wang, Robert McGibbon, Vijay S. Pande, and Todd J. Martínez, Automated discovery and refinement of reactive molecular dynamics pathways, Journal of Chemical Theory and Computation, 12, 638-649, 2015. [link]
- Jason Ford, Stefan Seritan, Xiaolei Zhu, Michael N. Sakano, Md Mahbub Islam, Alejandro Strachan, and Todd J. Martínez, Nitromethane decomposition via automated reaction discovery and an initio corrected kinetic model, The Journal of Physical Chemistry A, 125, 1447-1460, 2021. [link]
- Rui Xu, Jan Meisner, Alexander M. Chang, Keiran Thompson, and Todd J. Martínez, First principles reaction discovery: From the Schrodinger Equation to experimental prediction for methane pyrolysis, Chemical Science, 14, 7447-7464, 2023. [link]
- Alexander M. Chang, Jan Meisner, Rui Xu, and Todd J. Martínez, Efficient acceleration of reaction discovery in the ab initio nanoreactor: Phenyl radical oxidation chemistry, The Journal of Physical Chemistry A, 127, 9580-9589, 2024. [link]